
Prindërit e marrin shumë seriozisht rolin e tyre mbrojtës, edhe para se foshja e tyre të shohë për herë të parë dritën e botës. Çfarëdo problemi me të cilin përballet fëmija, prindi pranon detyrën – ta konstatojë shkakun e problemit dhe ta gjejë zgjidhjen. Prandaj, është shumë e vështirë për nënat dhe baballarët kur një problem i madh e shqetëson fëmijën e tyre, kurse nuk ka fajtor të vërtetë dhe asnjë zgjidhje adekuate.
Problemet shëndetësore me të cilat mund të përballet fëmija kur është ende në barkun e nënës, ndonjëherë shfaqen pa ndonjë shkak të njohur, nuk ka trajtim efektiv dhe pasojat mund të jenë shumë të rënda ose fatale. Ndarja e informacioneve të rëndësishme për çrregullimet kromozomale është shumë e rëndësishme, si për mjekët, ashtu edhe për prindërit, sepse ekziston qëllimi i përbashkët: gjetja e një zgjidhjeje në formën e parandalimit apo diagnostikimit të hershëm.
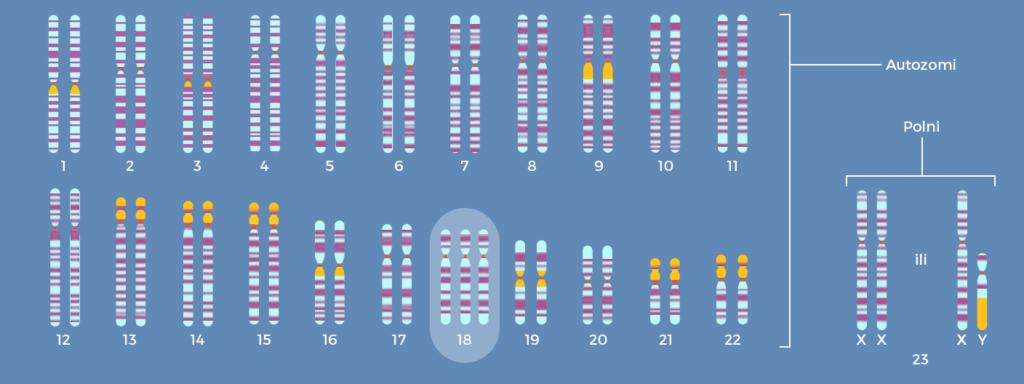
Si ndodh trisomia 18 (sindroma Edwards)?
Një qenie tipike njerëzore ka 46 kromozome, të organizuara në 23 çifte, të cilat trashëgohen nga prindërit. Në mënyrë që nga dy prindër të shëndetshëm biologjikisht të krijohet një fëmijë biologjikisht të shëndetshëm, kopjimi i materialit gjenetik duhet të bëhet pa gabime. Çdo gabim në kopjim mund të rezultojë në devijime serioze në krijimin e gjenotipit të një qenie të re, gjë që mund të rezultojë në probleme të ndryshme shëndetësore ose anomali fizike dhe intelektuale.

Prandaj, në mënyrë që fetusi të zhvillohet normalisht që nga momenti i fekondimit, kusht është kopjimi preciz i materialit gjenetik të prindërve. Njeriu kanë 23 çifte kromozomesh, të madhësive dhe formave të ndryshme, të gjitha të numëruara me kujdes sipas konventës ndërkombëtare. 22 çiftet e para i quajmë autosomale, ndërsa çifti i fundit përcakton gjininë e fëmijës. Çfarëdo devijim në kopjimin e materialit gjenetik në çifte kromozomesh do të rezultojë në sëmundje ose çrregullime të ndryshme. Gabimet në kopjimin e materialit gjenetik ndodhin në shtatzëninë shumë të hershme – ndonjëherë gjatë bashkimit të spermatozoidit dhe qelizës vezë dhe ndonjëherë në fazat e hershme të zhvillimit të embrionit. Një kopje shtesë e materialit gjenetik (3 në vend të 2 kopjeve) do të çojë deri te trisomia, ndërsa mungesa e materialit gjenetik (1 në vend të 2 kopjeve) do të çojë deri te monosomia. Sindroma Edwards ose trisomia 18, së bashku me sindromën Down dhe sindromën Patau, është një nga trisomitë më të zakonshme tek njerëzit.
Sindroma Edwards ose trisomia 18 ndodh (siç e tregon edhe vetë emri) kur një kopje shtesë e materialit gjenetik shfaqet në kromozomin e 18-të. Emrin e mban sipas gjenetistit anglez Edwards (John Hilton Edwards), i cili e përshkroi për herë të parë në vitin 1960 sindromën.
Incidenca dhe statistikat e sindromës Edwards

Sindroma Down është e vetmja trisomi që shfaqet më shpesh te njerëzit sesa sindroma Edwards. Incidenca e sindromës Edwards është 1:6000 foshnja të porsalindura. Megjithatë, simptomat janë shumë më serioze dhe prognoza shumë më e keqe te fëmijët e prekur nga trisomia 18 sesa tek ata që janë të prekur nga trisomia 21 (sindroma Down). Rezultatet e hulumtimeve tregojnë se nënat mbi 35 vjeç kanë një rrezik më të lartë për të lindur një fëmijë me trisomi 18 (si dhe me sindromën Down dhe Patau).
Në shumicën e rasteve (rreth 65%) shtatzënia e konstatuar me këtë patologji kromozomale përfundon me abort spontan, ndërsa zhvillimi i fetusit përfundon me vdekje intrauterine. Shumica e fëmijëve të lindur të gjalla (rreth 80%) janë vajza. Foshnjat që arrijnë të lindin zakonisht kanë probleme serioze shëndetësore dhe një pasqyrë klinike shumë të rënduar. Vetëm gjysma e fëmijëve të lindur me sindromën Edwards mbijetojnë dy muajt e parë pas lindjes, ndërsa vetëm 5-10% arrijnë të jetojnë deri në ditëlindjen e tyre të parë. Më pak se 1% jetojnë deri në vitin e dhjetë të jetës.
A mund të kenë prindërit e shëndetshëm një fëmijë me sindromën Edwards?
Prindër plotësisht të shëndetshëm, me një anamnezë familjare të mirë, mund ta kenë një fëmijë të prekur nga sindroma Edwards. Sindroma shfaqet si rezultat i bashkimit të qelizave riprodhuese me një numër jonormal të kromozomeve. Pavarësisht nëse qeliza vezë ose spermatozoidi mbartin gabimin gjenetik – embrioni do të ketë një kopje shtesë të kromozomit. Prandaj, trisomia 18 konsiderohet të jetë një rezultat i padëshiruar i lotarisë gjenetike.
Në një përqindje të vogël shfaqet mozaicizmi. Kjo do të thotë se disa qeliza mbeten tipike, ndërsa të tjerat preken nga trisomia. Tek mozaicizmi simptomat janë përgjithësisht më pak të theksuara, ndërsa pasqyra klinike është disi më e favorshme. Gjithashtu është i mundur edhe translokimi, kur materiali i tepërt i kromozomit 18 lidhet me një kromozom tjetër. Trisomia me translokim të ekuilibruar është e trashëgueshme dhe atëherë trisominë që e prek embrionin mund ta ndërlidhim me një translokim asimptomatik tek njëri prindër. Prindi nuk ka tepricë të materialit gjenetik, ka një fenotip normal dhe nuk ka simptoma të sëmundjes. Serioziteti i gjendjes shëndetësore tek mozaicizmi dhe translokimi përcaktohet individualisht dhe varet nga rasti në rast. Megjithatë, në shumicën e rasteve bëhet fjalë për një trisomi 18 të plotë, e cila ka simptoma karakteristike dhe e rëndon pasqyrën klinike në mënyrë të pritshme.
Simptomat, pasqyra klinike dhe prognoza e sindromës Edwards
Një fëmijë i prekur nga sindroma Edwards ka një pasqyre klinike shumë të rënduar dhe prognoza është e keqe. Nëse diagnoza nuk është konstatuar in vitro (në mitër), sindroma diagnostikohet lehtësisht që në lindje, duke u bazuar në anomalitë karakteristike fizike: pesha e vogël në lindje, koka me formë të panatyrshme, goja e vogël, buza dhe qiellza e çarë, veshët e ulët, të vegjël dhe mjekra e pazhvilluar, sy të vegjël dhe qepalla të ulura përgjysmë poshtë syve, gishta të shtrënguar në grushte dhe të mbivendosur, gishtat e mëdhenj dhe thonjtë të pazhvilluar.
Diagnoza konfirmohet më vonë me përcaktimin e kariotipit (përcaktimi i numrit të kromozomeve). Analiza laboratorike e gjakut të të porsalindurit mund të tregojë nivele të ulëta të trombociteve, qelizave të bardha të gjakut, anemi dhe policitemi (rritje e numrit të qelizave të kuqe të gjakut). Një ekzaminim me ultratinguj mund të tregojë ndryshime strukturore në zemër, si dhe keqformime të sistemeve të tjera të organeve. Me inçizim rentgen do të zbulohen anomalitë e mundshme në formimin e kockave.
Pra, problemet nuk ndalen vetëm në pamjen fizike. Janë të prekur sistemet e shumta organesh, si dhe inteligjenca. Menjëherë pas lindjes zbulohen anomalitë e zemrës dhe veshkave, ndërsa sindroma shpesh prek edhe traktin gastrointestinal. Rreth 90% e pacientëve kanë defekte serioze të zemrës. Përgjithësisht frymëmarrja është e vështirësuar për shkak të zhvillimit të pamjaftueshëm të mushkërive. Trisomia 18 prek edhe organet riprodhuese: testikujt e pazhvilluar dhe penisin e vogël tek djemtë dhe vezoret dhe labitë e pazhvilluara tek vajzat. Sindroma Edwards shoqërohet me mikrocefalinë (truri i vogël), hidrocefalinë (akumulimi i lëngjeve rreth trurit) dhe anencefalinë (truri i pazhvilluar). Prapambetja mendore është evidente, siç është edhe zhvillimi i ngadaltë motorik.
A ka kurë për sindromën Edwards?
Nuk ka kurë për sindromën Edwards, kështu që kujdesi është kryesisht paliativ dhe simptomatik. Qëllimi kryesor i kujdesit është t´i lehtësojë shqetësimet dhe dhimbjet dhe ta përmirësojë cilësinë e jetës. Siç kemi theksuar më lart, vetëm 1% e fëmijëve të prekur nga kjo patologji kromozomale jetojnë deri në ditëlindjen e tyre të dhjetë. Në këtë periudhë të shkurtër janë të mundshme ndërhyrjet e shumta kirurgjikale në mënyrë që të sanohen problemet me zemrën apo traktit tretës. Fëmijët do të jenë në kontakt të vazhdueshëm me pediatrin, kardiologun, kardiokirurgun, ortopedin, okulistin, nefrologun dhe ekspertët e tjerë.
Numri i qelizave të bardha të gjakut tek pacientët me sindromën Edwards është shpesh shumë i ulët, gjë që mund të çojë deri tek shfaqja e sepsës, e cila shpesh ka një përfundim fatal. Edhe fëmijët që jetojnë më shumë se 1 vit nuk do të jenë kurrë të pavarur – për shkak të prapambetjes mendore, zhvillimit të pamjaftueshëm motorik dhe problemeve të vazhdueshme shëndetësore. Në anën tjetër, për pavarësinë e fëmijëve të prekur nga trisomia 18 rrallë diskutohet, sepse jetëgjatësia mesatare është vetëm 5-10 ditë.
Diagnoza e hershme e sindromës Edwards
Në të kaluarën jo aq të largët, nuk ka ekzistuar mundësia e diagnostifikimit të hershëm të trisomisë – in utero (në mitër). Kjo do të thotë se diagnoza ishte e mundur vetëm pas lindjes dhe shpesh diagnoza do të ishte një tronditje për prindërit e rinj, të cilët prisnin një fëmijë të shëndetshëm. Kur dëgjojnë për herë të parë për një sëmundje që e prek fëmijën e tyre të porsalindur, prindërit interesohen menjëherë për prognozën dhe mundësinë e shërimit. Në rastin e një diagnoze të sindromës Edwards, mjekët, për fat të keq, nuk mund t´i inkurajojnë ose ngushëllojnë prindërit.
Meqenëse nuk ka kurë – alternativa tjetër më e mirë është diagnoza e hershme. Gjatë testimit prenatal analizohet një sasi e vogël e gjakut venoz të nënës dhe në këtë mënyrë merren rezultate mbi gjendjen shëndetësore të fetusit. Përkatësisht, përdoret metoda e veçantë për zbulimin dhe testimin e ADN-së së fetusit (cffDNA) nga gjaku i nënës. Testi klasik prenatal mund ta zbulojë praninë e trisomive më të zakonshme njerëzore (sindroma Down, Edwards dhe Patau), si dhe gjininë e foshnjës. Testet prenatale jo-invazive Premium Genetics (Premium Genetics monogjenik dhe Premium Genetics prenatal test), analizojnë regjione të dyshimta deri në dhjetë herë më shumë se testet e tjera që janë të disponueshme momentalisht në treg.
Premium Genetics monogjenik zbulon 100 sëmundje monogjene, 8 aneuploidi, 4 sindroma të mikrodelecionit dhe gjininë e foshnjës. Testi prenatal Premium Genetics mund ta zbulojë aneuploidinë autosomale, aneuploidinë e kromozomit seksual, mikrodelecionet dhe gjininë e fetusit. Mund të përdoret edhe tek shtatzënitë binjake.Testet janë të disponueshme për të gjitha gratë shtatzëna që nga java e dhjetë e shtatzënisë. Bëhet fjalë për testet që janë të shpejta, të sakta, të besueshme dhe të sigurta si për nënën, ashtu edhe për fëmijën. Nëse testi prenatal tregon praninë e mundshme të anomalive kromozomale, gruaja shtatzënë i referohet më tej procedurave diagnostike invazive, siç është amniocenteza, e cila mbart disa rreziqe.
Rëndësia e testimit prenatal
Të dhënat statistikore tregojnë se mbi 90% e shtatzënive në Evropë ndërpriten nëse prania e anomalive kromozomale zbulohet herët në shtatzani. Konsiderojmë se është shumë e rëndësishme që prindërit të jenë në gjendje ta bëjnë një zgjedhje dhe informohen në kohë, në mënyrë që të marrin me qetësi një vendim për hapat e mëtejshëm. Diagnoza e sindromës Edwards nuk sjell asnjë grimë optimizmi – përkundrazi, lajmet e këqija rreshtohen njëri pas tjetrit. Nuk ka asnjë shpresë për përmirësim apo shërim. Testimi prenatal u mundëson prindërve të ardhshëm që t´i heqin qafe shqetësimet më të vështira në një shtatzëni shumë të hershme dhe kështu të vazhdojnë ta presin me padurim takimin e parë me fëmijën e tyre. Rezultatet e testit që konfirmojnë shëndetin e fëmijës do të sjellin paqe dhe qetësi në çdo shtëpi.