Çfarë është
Testi Premium Genetics?
Testi Premium Genetics është test prenatal jo-invaziv (NIPT) për zbulimin e anomalive kromozomale fetale, i aplikueshëm për shtatzënitë teke, shtatzënitë me binjakë dhe shtatzënitë me binjak të humbur.
Testi përdor teknologji unike, vetanake, të bazuar në kërkimet dhe zhvillimet e fundit në gjenetikën molekulare dhe diagnostikën.

Testin Premium Genetics e ka zhvilluar një ekip shkencor me mbi 25 vjet përvojë në fushën e diagnostifikimit prenatal, mjekësisë molekulare, gjenomikës, transkriptomikës, metilomikës dhe bioinformatikës.

Testi Premium Genetics përdor teknologji të re të targetimit, e cila mundëson zbulimin superior të aneuploidive dhe saktësinë e pakrahasueshme të matjes së fraksionit fetal. Regjionet e synuara në kromozomet 21, 18, 13, X dhe Y u lokalizuan, u zmadhuan dhe u analizuan për aneuploidi, duke përdorur teknologjitë tona gjenetike dhe bioinformatike.

Testi Premium Genetics i tejkalon kufizimet e teknologjive aktualisht të disponueshme të testimit prenatal, duke përdorur metodën më të avancuar të leximit të thellë, përkatësisht duke i kontrolluar regjionet deri në 10 herë më shumë se testet e tjera.
Trisomitë

SINDROMA DOWN
Kopja shtesë e kromozomit 21
Frekuenca: 1 në 700
Karakteristikat themelore janë: lartësia më e shkurtër e trupit, sytë në formë bajame të cilat janë të vendosura në mënyrë të pjerrët, njollat e Brushfield – të bardha, pikat në formë të gjysmëhënës në bebëzat, llapat e vogla të veshit të vendosura më poshtë, gjuha më e madhe që del nga zgavra e gojës, çrregullimet e gjoksit, dermatoglifët karakteristike. Shkalla e prapambetjes mendore mund të jetë e shkallëve të ndryshme. Ekstremitetet janë më të shkurtra me vijën karakteristike të katër gishtave. Defektet kongjenitale të zemrës janë të pranishme në 50% të rasteve.

SINDROMA EDWARS
Kopja shtesë e kromozomit 18
Frekuenca: 1 në 5.000
Karakteristikat themelore janë: pesha më e vogël e trupit në lindje, forma jotipike e kokës, llapat e vogla të veshit, epikantusi (palosje e lëkurës që mbulon këndin e brendshëm të syrit), çarje e buzës dhe qiellzës, nofulla e vogël. Një gamë e gjerë ndryshimesh prek ekstremitetet, më të zakonshmet prej të cilave janë grushti i shtrënguar dhe pozita tipike e gishtërinjve, ku gishti i dytë kalon mbi të tretin, ndërsa i pesti mbi të katërtin. Sindroma shoqërohet me defekte të rënda të zemrës, anomali të sistemit të tretjes dhe të veshkave. Kryesisht, ata vdesin në periudhën neonatale.

SINDROMA PATAU
Kopja shtesë e kromozomit 13
Frekuenca: 1 në 16.000
Në mënyrë intrauterine mund të vërehen vonesat në rritje, si dhe pesha e vogël gjatë lindjes. Sindroma shoqërohet me një formë dhe madhësi specifike të kokës (kokë e vogël), defekte të fytyrës, siç është çarja e buzës, nofullës dhe qiellzës, mikroftalmi (moszhvillimi i syve). Holoprosencefalia (truri nuk është i ndarë në dy hemisfera) është një gjetje e shpeshtë. Mund të vërehen defekte të murit të barkut, anomali të rënda të zemrës, toni i dobësuar i muskujve, anomali të ndryshme të duarve dhe këmbëve (polidaktilia). Shumica mbijetojnë vetëm ditët e para pas lindjes.
Aneuploidi kromozomale të gjinisë

SINDROMA TURNER
Mungesa e një kromozomi X
Frekuenca: 1 në 2.000 tek fëmijët femra
Periudha e foshnjës karakterizohet, përveç peshës së vogël gjatë lindjes, me një qafë të gjerë dhe një vijë të ulët flokësh, pozitë jonormale të llapave të veshit, qiellzë të lartë, limfedemë të këmbëve dhe të duarve, e cila përgjithësisht zhduket gjatë vitit të parë të jetës. Koarktacioni i aortës është defekti më i zakonshëm i zemrës. Në moshën e mëvonshme të jetës mungojnë karakteristikat sekondare gjinore, ato rrallë mbeten fertile ose nëse mbeten, kanë një rrezik të shtuar të abortit për shkak të anomalive kromozomale të fetusit.

SINDROMA TRIPLE X
“FEMRA E FORTË”
Kopja shtesë e kromozomit X
Frekuenca: 1 në 1.000 tek fëmijët femra
Vërehet rritja e lartë, epikantusi, klinodaktilia e gishtit V (devijimi i gishtit të vogël), me prapambetje mendore më të lehtë. Në një numër më të vogël të rasteve shfaqet oligomenorrhea dhe menopauza e parakohshme. Ekziston rreziku nga problemet psikiatrike, veçanërisht skizofrenia. Shumica e personave lindin fëmijë me një kariotip normal.

SINDROMA KLINEFELTER
Kopja shtesë e kromozomit X
Frekuenca: 1 në 1.000 tek fëmijët meshkuj
Zhvillimi fizik i djemve nuk dallon deri në moshën 4 vjeçare. Bëhet fjalë për djem me testikuj të vegjël dhe të fortë, të cilët kanë probleme në shkollë, maturohen më vonë mendërisht dhe emocionalisht, janë shumë të ndjeshëm dhe të pambrojtur. Në moshën e rritur, ata janë të gjatë dhe kanë muskulaturë më pak të zhvilluar. Mund të jetë e pranishme prapambetja e lehtë mendore, e cila reflektohet në sferën e shprehjes së të folurit. Pothuajse të gjithë janë infertilë. Kanë një rrezik të shtuar ndaj zhvillimit të kancerit të gjirit dhe skizofrenisë.

SINDROMA JACOBS
Kopja shtesë e kromozomit Y
Frekuenca: 1 në 1.000 tek fëmijët meshkuj
Pasqyra klinike karakterizohet nga anomali të tilla, siç janë: llapat e zhvilluara dobët të veshëve, anomalitë e gjoksit, klinodaktilia e gishtit V (devijimi i gishtit të vogël), hernia inguinale. Gjithashtu, ekziston edhe delikuenca e hershme, e cila shfaqet rreth moshës 6-vjeçare me gënjeshtra, vjedhje, konflikte të shpeshta në shkollë. Personat në fjalë nuk përshtaten mirë shoqërisht, por me arsim adekuat, ata mund të udhëhiqen siç duhet. Fertiliteti është i ruajtur.

SINDROMA XXYY
Kopja shtesë e kromozomit X dhe Y
Frekuenca: 1 në 17.000 tek fëmijët meshkuj
Disa shenja dhe simptoma të kësaj gjendje përfshijnë: vonesa në zhvillim, si dhe vështirësi në të mësuar dhe dëmtime të të folurit. Shpesh është i pranishëm edhe mungesa e vëmendjes e shoqëruar me disa aftësi të kufizuara intelektuale. Vërehet hiperaktiviteti, siç janë çrregullimet nga spektri i autizmit. Kryesisht bëhet fjalë për personat e gjatë me çrregullime të funksionit riprodhues.
Mikrodelecione (edhe për shtatzënitë me binjakë)

SINDROMA DI-GEORGE
Mungesa e një pjese të kromozomit 22
Frekuenca: 1 në 1.000
Karakterizohet nga anomali kongjenitale të zemrës, mungesë ose hipoplazi e timusit (me mungesë imuniteti dhe infeksione të shpeshta), hipoparatiroidizëm me pasojë hipokalceminë dhe çrregullime të aparatit të tretjes. Zhvillimi psikomotor është i ngadalësuar. Ata janë të prirur për zhvillimin e çrregullimeve psikiatrike dhe krizave të vetëdijes. Vdekshmëria është e lartë gjatë vitit të parë pas lindjes, kryesisht për shkak të defekteve të zemrës. Nëse arrijnë moshën madhore, kërkojnë një kujdes dhe mbikëqyrje të vazhdueshme.

SINDROMA MIKRODELECIONE
Mungesa e e një pjese të kromozomit 1
Frekuenca: 1 në 5.000
Zakonisht shkakton paaftësi të rëndë intelektuale me çrregullime të sjelljes. Më shumë se gjysma e të sëmuarëve kanë dëmtime të rënda të trurit të shoqëruara me kriza të vetëdijes. Shpesh vërehen hipotoni (tonusi i dobët i muskujve) dhe vështirësi gjatë gëlltitjes. Kanë kokë të vogël me tipare karakteristike të fytyrës (kokërdhokët e syve të fryrë, vetulla të rrafshuara, pjesa qendrore më pak e zhvilluar e fytyrës, hundë e gjerë dhe e sheshtë, veshë të vendosur poshtë dhe me formë të çrregullt). Personat me Sindromën e fshirjes 1p36 mund të kenë probleme me shikimin ose dëgjimin.

SINDROMA SMITH MAGENIS
Mungesa e një pjesë të kromozomit 17
Frekuenca: 1:15.000
Personat e prekur nga sindroma Smith Magenis zakonisht kanë një fytyrë karakteristike të gjerë, sy të vendosur thellë, tonus të ulët të muskujve dhe gjatësi të shkurtër. Ndonjëherë shfaqet edhe skolioza. Kryesisht është e pranishme humbja e plotë ose e pjesshme e dëgjimit, shkëputja e retinës, por gjithashtu mund të shfaqen edhe problemet me veshkat dhe zemrën. Çrregullimet intelektuale janë të shpeshta.

SINDROMA WOLF HIRSCHHORN
Mungesa e një pjesë të kromozomit 4
Frekuenca: 1.50.000
Karakteristikat fizike të qarta tregojnë sindromën Wolf Hirschhorn: koka është e vogël dhe me formë të çrregullt, hunda është e sheshtë, balli është i lartë, distanca midis syve është e theksuar. Koka mund të jetë asimetrike dhe mund të vërehen edhe keqformime të veshëve dhe probleme me dëgjimin. Organet e brendshme si zemra, truri dhe veshkat janë gjithashtu të prekutra. Shfaqet edhe prapambetja mendore. Fëmija ka tonus të ulët të muskujve, përparon më ngadalë dhe mbetet prapa në zhvillimin e aftësive motorike.
Analiza gjenomike e synuar
Testet prenatale Premium Genetics përdorin sekuenca vetanake, të synuara dhe të dizajnuara për t´i shmangur sekuencat me numër variabil të kopjeve (CNV), elementët e përsëritur të ADN-së dhe arkitekturën komplekse të gjenomit. Kjo qasje e synuar i tejkalon problemet që ndërlidhen me NIPT-et e tjera dhe rrit saktësinë e këtij testi. Kjo sjell rezultate më pak false pozitive.
Thellësia e sekuencimit dhe saktësia e leximit
Thellësia e leximit është numri i herëve gjatë të cilëve lexohet nukleotidi në gjenomin gjatë analizës. Testi Premium Genetics i regjistron fragmented e ADN-së vetëm nga regjionet e synuara të kromozomeve të interesit. Në këtë mënyrë, është në gjendje t´i lexojë regjionet e selektuara në një thellësi jashtëzakonisht të lartë të leximit, gjë që përmirëson saktësinë statistikore të analizave dhe rrit ndjeshmërinë dhe specifikën e Testit Premium Genetics.
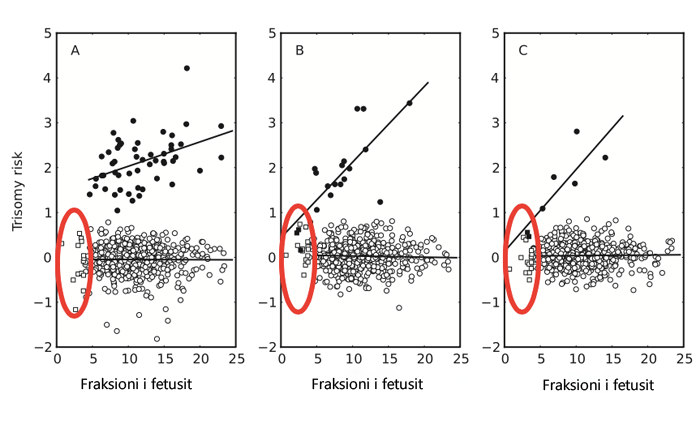
Matja e fraksionit fetal
Testi Premium Genetics përdor lokacione informuese për të dalluar me besueshmëri cfDNA fetale nga cfDNA e nënës. Softueri i bioinformatikës i patentuar i përdor pikat e thellësisë së lartë të leximit të këtyre lokacioneve informative për të llogaritur me saktësi fraksionin fetal. Masat precize të fraksionit fetal e rrisin saktësinë dhe besueshmërinë e Testit Premium Genetics.
Testi ynë prenatal mat me saktësi FRAKSIONIN FETAL
Ndryshe nga testet e tjera që vlerësojnë fraksionin fetal, testet prenatale Premium Genetics, falë teknologjisë më të avancuar të patentuar, matin saktësisht pëqindjen e ADN-së së foshnjës në gjakun e nënës. Kjo është jashtëzakonisht e rëndësishme, sepse një vlerësim i dobët i përqindjes së ADN-së së foshnjës çon deri tek rezultatet jo të besueshme, ndërsa me matje të sakta dhe raportim të sigurtë të ADN-së së foshnjës, ne sigurojmë një përqindje më të ulët të rezultateve false pozitive dhe një besueshmëri prej 99,99%.
TESTI MË GJITHËPËRFSHIRËS PRENATAL NË BOTË
- Deri në 229 sëmundje monogjene
- 8 aneuplodi
- 4 sindroma mikrodelecione
- Gjininë e foshnjës
Analiza krahasuese e testimit prenatal
Saktësia e Testit Premium Genetics në krahasim me testet e tjera
Siguria e Testit Premium Genetics në krahasim me testet e tjera
Rritja e thellësisë së leximit në krahasim me testet e tjera
